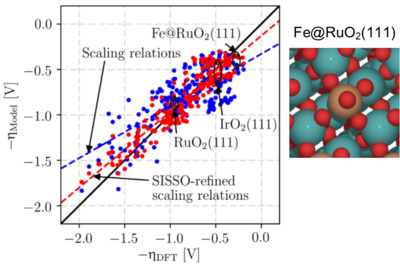
Computational screening of transition metal oxide catalysts is challenging due to their more localized and intricate electronic structure as compared to transition metal catalysts and the resulting lack of suitable activity descriptors to replace expensive density-functional theory (DFT) calculations. In recent work we used a compressed sensing approach to identify descriptors in the form of algebraic expressions of surface-derived features for predicting adsorption enthalpies of oxygen evolution reaction (OER) intermediates at doped RuO2 and IrO2 electrocatalysts. Our descriptors significantly outperform previously highlighted single descriptors both in terms of accuracy and computational cost. Compared to standard scaling relations that employ the oxygen adsorption enthalpy as unique reactivity descriptor, our analysis reveals that the consideration of features related to the local charge transfer leads to significantly improved refined scaling relations. These allow us to screen for improved OER electrocatalysts with an uncertainty in the theoretical overpotential similar to the expected intrinsic DFT error of 0.2 V.
Read more about our work:
