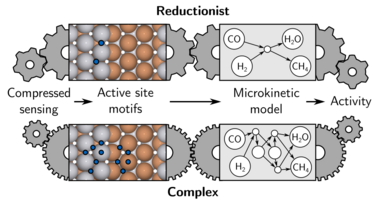
Computational screening based on first-principles microkinetic modeling has evolved into a widespread tool for catalyst discovery. Efficiently exploiting various scaling relations, this approach draws its predictive character from reliable adsorption energies, typically calculated with density-functional theory (DFT). In prevalent screening approaches the concomitant computational costs are kept tractable through the use of reductionist microkinetic models that only resolve a minimalistic amount of active site motifs at the catalyst surface. In recent works, we have argued that the uncertainty arising from the employed active site representation is one of the major bottlenecks towards an ever extended and reliable use of such computational screening for catalyst discovery. We have scrutinized common practices by systematically comparing the screening predictions for the CO methanation reaction when using microkinetic models that resolve an increasing amount of sites, up to the full consideration of all high-symmetry sites at stepped transition metal (TM) and binary TM alloy catalysts. Apart from generally overestimating the catalytic activity, the simplified models fail to identify a most promising class of layered bimetallic alloys as their insufficient representation of the catalyst surface does not allow them to correctly capture the rate-determining step. We further argue that with the current methodological advances in areas such as compressed sensing and machine learning, and the concurrent availability of cheap adsorption energetics for a wide range of possible catalyst materials, there is no reason to continue to use simplistic microkinetic models in computational catalyst screening.
Read more about our work:
