I obtained my PhD degree in 2014 from Aarhus University under the supervision of Prof. Bjørk Hammer and Prof. Liv Hornekær. During my PhD studies, I researched graphene and polycyclic aromatic hydrocarbons (PAHs) using theoretical modeling in the form of density functional theory (DFT). I worked closely together with experimental partners, in particular from the group of Prof. Liv Hornekær. The topics covered in my thesis include hydrogenation of PAHs and their catalytic role for H2 formation in interstellar space, how to control the electronic properties of graphene by reacting it with hydrogen or by inserting atoms or molecules between the graphene sheet and the underlying substrate, and the efficiency of a graphene coating towards the penetration of atoms and small molecules.
In 2014 I joined the group of Prof. Karsten Reuter at the Technical University of Munich (TUM), first as an Alexander von Humboldt postdoctoral fellow and from 2017 as a group leader. In the last couple of years, I have been building up a group at TUM working on heterogeneous catalysis. Apart from DFT and various semi-empirical methods, my group and I apply non-equilibrium statistical mechanics techniques such as kinetic Monte Carlo (kMC), mean field-based microkinetic modeling, ab initio thermodynamics as well as machine learning (ML) and compressed sensing methods. We also focus on method development and are co-developers of the kMC code “kmos”. Among other things, we apply these methods to studying the reactivity of metal and oxide catalysts, bifunctional catalysis, and graphene growth at liquid metal catalysts.

1520-332 Ny Munkegade 120
DK - 8000 Aarhus C
mie@phys.au.dk
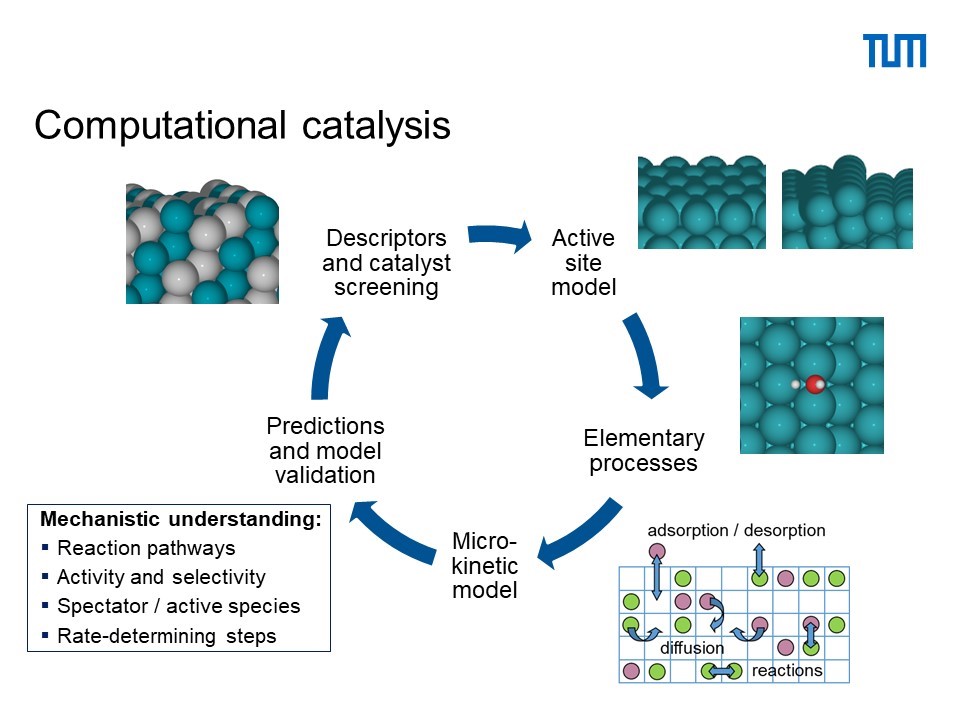
During my AIAS Fellowship I will work on developing and applying theoretical methods for describing and predicting the complex reaction networks and active catalyst materials involved in the formation of complex organic molecules at dust grains in interstellar space. The materials complexity, together with the combinatorial explosion of possible reaction pathways leading to the formation of larger organic molecules, renders the construction of a detailed microkinetic model challenging. In particular, the direct calculation of model inputs with expensive first-principles calculations such as DFT becomes impractical. The objective of this project is to identify descriptors that allow for low-cost prediction of required input (e.g. adsorption energies, reaction barriers) to a microkinetic model of dust grain reactivity. The descriptors will be formulated as non-linear functions of a set of descriptive parameters, e.g. bond distances and angles or tabulated atomic or bulk properties of the elements making up the active site of the dust grain. The recently developed compressed sensing method, SISSO, will then be used to select the descriptors that provide the best predictions of DFT-calculated model inputs. The resulting screening model will be iteratively refined and used to search for promising active site motifs. The experimental investigations from InterCat partners and the theoretical work will complement and guide each other. For example, the theoretical prediction of promising dust grain catalysts could subsequently be tested experimentally. Or the experimental discovery of specific reaction intermediates could be added to the theoretical reaction network investigated.