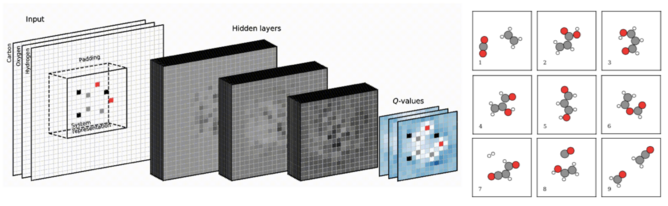
Modern simulation techniques such as density functional theory (DFT) allow for an accurate quantum mechanical description of matter. This means that given the atomistic structure, the stability and physico-chemical properties of e.g. molecules, nano clusters, and crystals may be calculated. However, the atomistic structures involved are often assumed, inferred from experiment or simply guessed. In the group we strive to develop and employ methods that automate the structure determination. The methods build extensively on machine learning techniques, which decrease the computational time scales involved in global optimization of atomistic structure. The aim of the work is to develop artificially intelligent systems that may design matter with desired properties.
To learn more about the projects going on in the group:
http://phys.au.dk/~hammer/machine_learning_projects.html
