The aim of our research is to understand and tailor the catalytic properties of materials at the atomic scale using computational modelling and machine learning. Applications cover heterogeneous catalysis and catalytic processes at the surfaces of dust grains in the interstellar space.
Materials with a catalytic function may be found in such diverse places as chemical reactors or as dust grains in molecular clouds in the interstellar space. Despite their crucial role in society and the Universe for accelerating chemical reactions, the reliable description of catalytic properties and the prediction of what materials may be even better catalysts than those we know already is still challenging. While it is possible to use quantum mechanical calculations (density functional theory, DFT) to study simple reactions and simple model catalysts, the computational demands of such an approach can quickly become prohibitively large for realistic materials and reaction networks.
In the group we use various computational modelling and simulation tools based on quantum mechanics, statistical physics and machine learning to study catalytic reactions at different kinds of surfaces. We develop relationships between the composition and structure of the surface and the catalytic activity and selectivity to specific reaction products, including detailed mechanistic understanding about the dominant reaction pathways and rate-determining steps. As such, we work at the interface between physics and chemistry, with recent machine learning methods applied having their origin in computer science.
We will soon have openings for a PhD and a postdoc position. Stay tuned.
Students interested in doing their BSc or MSc projects in the group are invited to contact Mie Andersen.
Click on the images below to learn more about ongoing research projects

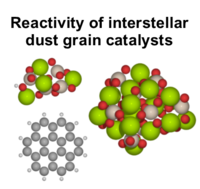
Luuk Kempen, Marius Juul Nielsen, Mie Andersen
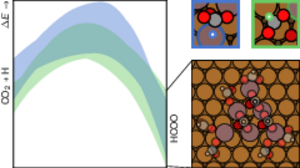
Niels M. Mikkelsen, Thanja Lamberts, Mie Andersen
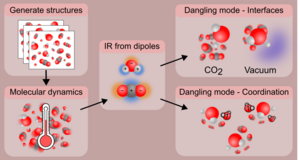
Luuk Kempen, Mie Andersen
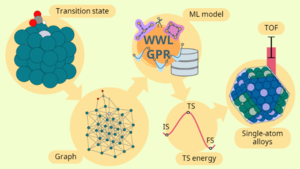
Raffaele Cheula, Mie Andersen

Raffaele Cheula, Thien An Michael Quoc Tran, Mie Andersen

Mie Andersen, Andreas Møller Slavensky
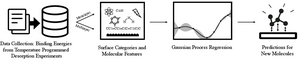
Torben Villadsen, Niels F.W. Ligterink, Mie Andersen
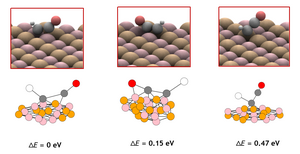
Wenbin Xu, Karsten Reuter, Mie Andersen
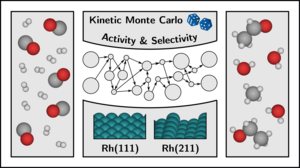
Martin Deimel, Hector Prats, Michael Seibt, Karsten Reuter, Mie Andersen
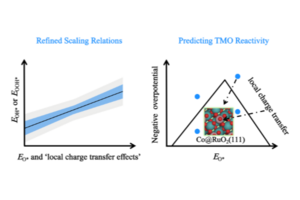
Wenbin Xu, Mie Andersen, Karsten Reuter
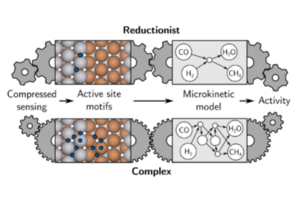
Martin Deimel, Karsten Reuter, Mie Andersen
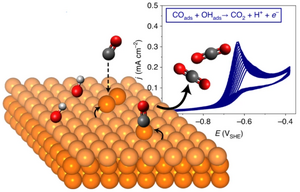
Andrea Auer, Mie Andersen, Eva-Maria Wernig, Nicolas G. Hörmann, Nico Buller, Karsten Reuter, Julia Kunze-Liebhäuser

Juan Santiago Cingolani, Martin Deimel, Simone Köcher, Christoph Scheurer, Karsten Reuter, Mie Andersen
